Apolipoprotein E4 (ApoE4) is the strongest common genetic risk factor for late-onset Alzheimer's disease, yet no approved therapy addresses it directly. We describe a multimodal machine-learning platform that spans the design arc — from a structurally anchored target through generative molecular design, physics-based screening, and multi-parameter safety modeling — in a single optimization loop. Applied to ApoE4, the platform generated and evaluated a large in-silico library against the only experimentally observed small-molecule binding site, yielding a series of lead structure correctors with strong predicted affinity and clean computational safety profiles. The lead series is protected by a filed provisional patent. Quantitative results are shared under NDA. All results to date are computational; experimental validation is the objective of the next phase. This public edition presents the problem, approach, and outcomes; proprietary methods are withheld and available to qualified partners under NDA.
1 · The problem
Alzheimer's disease affects an estimated 55 million people worldwide, projected to reach 139 million by 2050, at a global cost exceeding $1.3 trillion annually. Despite recent amyloid-directed approvals, the field lacks therapies that address the disease's strongest genetic driver.
That driver is ApoE4. Carrying a single copy raises late-onset Alzheimer's risk several-fold; carrying two copies raises it by roughly an order of magnitude. ApoE4 differs from the common, benign ApoE3 isoform by a single amino-acid substitution that promotes a pathogenic, misfolded conformation — a phenomenon known as domain interaction. Correcting that conformation with a small molecule (a "structure corrector") is a mechanistically grounded strategy with supporting cellular and genetic evidence.
The challenge is chemistry. ApoE is a large, flexible, predominantly helical protein with few obvious druggable pockets. For years the strategy lived largely in hypothesis — until an experimentally observed structural foothold appeared.
2 · Why now
Three developments converged to make this tractable:
- An observed binding site. A co-crystal structure of ApoE4 with a bound small molecule (PDB 6NCO / 6NCN) established, for the first time, an experimentally observed pocket suitable for structure-corrector design.
- Generative chemistry at scale. Modern generative models can propose novel, synthesizable molecules conditioned directly on a three-dimensional pocket, exploring chemical space far beyond enumerated libraries.
- Fast, physics-grounded evaluation. Structure-aware simulation now ranks candidates with enough fidelity — and speed — to drive a closed optimization loop rather than a one-shot screen.
The opportunity is the gap between overwhelming target validation and zero direct therapies. That gap is exactly where a computational platform built for hard targets creates value.
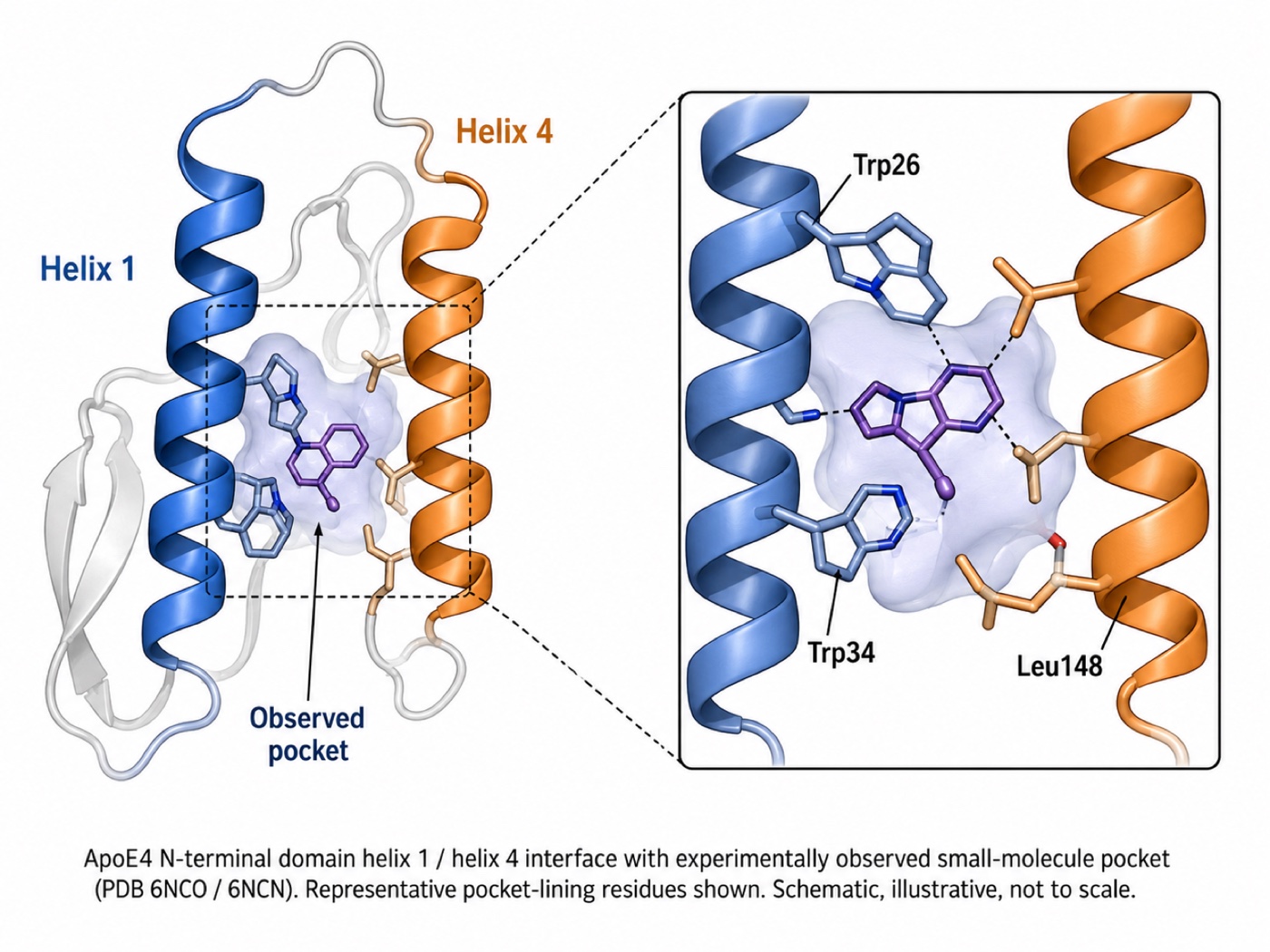
Figure 1 — The experimentally observed ApoE4 helix-1/4 pocket (PDB 6NCO / 6NCN) that Titan was designed against.
3 · The approach
Thea's platform treats discovery as a single multi-objective search rather than a linear hand-off. Generation, scoring, and safety modeling operate together, so each candidate is evaluated for potency, selectivity, and safety simultaneously. At a high level, the loop runs in five stages:
Discover
Generate
Screen
Optimize
Validate
Figure 2 — The closed discovery loop. Stages 01–04 are complete for the ApoE4 program; stage 05 (experimental validation) is the partner-funded next step.
Every program begins from a genetically validated target and an experimentally observed binding site — never a purely computational hypothesis. (Extending the platform upstream to discover novel targets is an active research direction; for ApoE4, the target and pocket come from human genetics and published crystallography, not from our platform.) We generate novel chemistry purpose-built for that pocket; rank it with physics-based, structure-aware scoring that we cross-check using independent methods; and gate every candidate through a full ADMET and structural-alert safety profile. An optimizer iterates the series toward the entire objective at once.
The architectures, training strategy, scoring formulation, optimizer configuration, and design rules that make this loop effective are proprietary. They are summarized in §6 and disclosed only under NDA.
4 · Results
Applied to ApoE4, the platform produced the following outcomes in its first campaign. Quantitative figures are reserved for the NDA edition; the qualitative results below are what they establish:
| Metric | Result |
|---|---|
| Library generated & screened | Large in-silico NDA |
| Binding site | Experimentally observed (6NCO / 6NCN) |
| Lead binding scores | Disclosed under NDA |
| Leads passing multi-parameter ADMET | Multiple |
| Structural alerts in final leads | None |
| Chemical novelty | Novel composition of matter |
| IP status | Provisional filed |
Figure 3 — The selection funnel. Absolute counts are reserved for the NDA edition; the shape conveys the discipline — a large generated library narrowed by physics, cross-method agreement, and a hard safety gate down to a small lead series.
| Property (anonymized) | Lead A | Lead B | Lead C |
|---|---|---|---|
| CNS MPO | |||
| BBB penetrance (pred.) | |||
| hERG safety | |||
| CYP liability | |||
| TPSA (in range) | |||
| MW (in range) | |||
| LogP (in range) | |||
| PAINS / BRENK alerts | Clean | Clean | Clean |
Figure 4 — Anonymized profiles for three representative leads across the CNS / safety axes used as gates. Directional ratings only; underlying numerical values (CNS MPO, BBB score, hERG, CYP, TPSA, MW, logP) are shared under NDA.
Top candidates were confirmed by independent scoring before acceptance — we treat convergence across methods, not any single number, as the bar for a result. The final leads were explicitly co-optimized for CNS drug-likeness (molecular weight, lipophilicity, polar surface area, predicted blood-brain-barrier penetrance) alongside affinity, so potency was not bought at the cost of developability.
5 · Validation & limitations
We are deliberate about what "validated" means. All results to date are computational predictions from physics-based and machine-learning models (the underlying figures are shared under NDA). They establish a strong, de-risked starting point — not a clinical claim.
The next phase, which we are seeking to fund through partnership, is experimental:
- Synthesis of lead candidates and biophysical confirmation of target engagement (e.g. SPR, ITC, co-crystallography).
- Cellular assays demonstrating functional structure correction in ApoE4 models.
- In-vitro ADME (microsomal stability, Caco-2 / MDCK-MDR1 permeability, plasma protein binding) and early in-vivo pharmacokinetics to confirm the predicted safety and exposure profile.
- Medicinal-chemistry optimization toward an IND-enabling development candidate.
Honest framing. Computational work reduces risk and cost; it does not replace the wet lab. Our claim is precise: a novel, patent-pending lead series — computationally nominated and cross-validated, ready for experimental confirmation.
Disclosed under NDA
- Exact molecule and lead counts
- Predicted binding affinities & scores
- Lead structures (SMILES) & composition-of-matter genus
- Full multi-parameter ADMET tables
- Model architecture, design rules & scoring
Not yet experimentally proven
- Synthesis of the lead candidates
- Biophysical binding (SPR / ITC)
- Co-crystal structure with ApoE4
- Cellular ApoE4 structure-correction
- In-vitro ADME and in-vivo PK
6 · What's protected
This public edition is intentionally silent on the mechanics that produce our results. The full edition, available under NDA, covers the model architectures and training approach, generative design rules, scoring and selection logic, optimizer configuration, the complete ADMET protocol, and the lead structures and composition-of-matter genus.
Methods — available under NDA
Section 3 (detailed), 4 (full data), and the methods appendix are reserved for qualified partners.
Architecture: redacted · Design rules: redacted · Scoring: redacted · Lead SMILES: redacted
References
- Petros A.M. et al. (2019). Fragment-based discovery of ApoE4 stabilizers. J. Med. Chem. 62:4120. (Co-crystal structures, PDB 6NCO / 6NCN.)
- Mahley R.W. & Huang Y. (2012). Apolipoprotein E structure correctors as therapeutics. J. Med. Chem.
- Chen H.-K. et al. (2012). Small-molecule structure correctors of ApoE4. J. Biol. Chem.
- Wang C. et al. (2018). iPSC-based validation of the structure-corrector approach. Nature Medicine.
© Thea Biosciences. Public edition. Forward-looking statements based on computational predictions; not medical advice.